Генетические изменения, обусловленные мутациями de novo. De novo мутации в гистон-модифицирующих генах как одна из общих причин генетических заболеваний Мутации de novo
Материал из BrainstormWiki
Что такое мутация?
Начнем с того, что “правильного” генокода не существует. Поэтому если у меня ген один, а у вас другой, то это не означает еще, что один из нас мутант.
Тем не менее, существуют такие изменения в генокоде, которые приводят к явным проблемам, и обычно они редко встречаются; их и называют мутации. Изменения, которые встречаются у более чем 1% населения, правильнее называть не мутации, а полиморфизмы .
Мутации можно унаследовать у родителей, или же они могут возникнуть только у ребенка - в этом случае их называют de novo
Мутации могут возникнуть уже при развитии организма, при делении и дифференциации клеток - такие называют соматическими мутациями ; их нельзя унаследовать, потому что они “живут” в клетках тела (сома), но не в половых клетках..
Наоборот, мутации, которые затрагивают половые клетки, наследуются, и их называют мутациями в зародышевой линии (germline mutations)
Мутации могут быть молчаливыми - они есть, а эффекта нет. Таких подавляющее большинство - если помните, гены занимают лишь менее 2% всей ДНК. Каждый из нас является носителем как минимум десятков таких мутаций. Если они приходятся на малозначимые участки ДНК, никто ничего не заметит.
Мутации могут затрагивать одну “букву” генокода - а могут и целый огромный фрагмент ДНК. Этот фрагмент чаще всего может быть потерян (делеция) или задвоен (дупликация) - в этом случае клетки изменят количество синтезируемых белков по затронутым генам - изменится их доза
Для разных видов мутаций существуют разные виды анализов и тестов, и мы по ним сейчас попробуем пройтись.
От больших - к маленьким
Анеуплодия - аномалия числа хромосом
Самая большая по размеру генетическая аномалия - изменение числа хромосом. Хотя это и не аутизм, но начать стоит с него 50 лет назад было открыто, что синдром Дауна вызывается тремя 21 хромосомами, вместо двух. Это называется трисомией; а изменение вообще числа хромосом - анеуплодией. Помимо этого встречается трисомия 13 хромосомы (синдром Патау) и 18 (синдром Эдвардса), а также ряд анеуплодий половых хромосом (X и Y)
Все это можно разглядеть в обычный оптический микроскоп - анализ называется “кариотипирование”. Хромосомы делящихся клеток фотографируют, сортируют и описывают. На картинке виден кариотип женщины с тремя 21 хромосомами.
Эффект от трисомий огромен и заметен в очень многих процессах: и в строении тела, и в физиологических процессах, и даже по характерным белкам в крови носящей матери (именно так работают сейчас скрининговые тесты на с. Дауна)
Почему встречаются только трисомии 21, 13 и 18 и половых хромосом? Случиться это может с любой хромосомой, но остальные не выживают даже до стадии заметной беременности. Возможно причина в том, что 21, 13 и 18 хромосомы - одни из самых “бедных” белковыми генами (помните, нумерация там по размеру, они ведь еще и маленькие) и увеличение их дозы относительно переносимо. Это подтверждается таблицей ниже: на ранних фазах развития возможны любые анеуплодии, а по мере приближения к успешным родам - только вот эти 3.

Copy Number Variation

CNV - это тоже либо задвоение, либо отсутствие определенной части ДНК, но уже не на уровне целой хромосомы, а несколько миллионов нуклеотидов.
Для их обнаружения применяют тестирование, называемое CGH array: comparative genomic hybridization, также FISH и другие.В нашей практике часто называют также “молекулярное кариотипирование”, микроматричный хромосомный анализ и т.п.
Это все равно огромные куски ДНК, которые могут включать в себя десятки генов и управляющих структур. Эффект от них очень заметен, почти всегда включает в себя дисморфизмы и измененения когнитивных способностей. Обозначают их по затронутому участку генокода, например 2q32 deletion - потеря участка в полоске 32 на длинной руке 2 хромосомы (вспоминаем часть 1 и “адреса” участков хромосом) Туда же относятся и многие известные “именные” синдромы, например синдром Вильямса - 7q11.23 deletion
Такие CNV мутации, как считается объясняют от 3% до 10% случаев аутизма. Это практически единственный вид генетической аномалии, который уверенно связан с некоторыми - синдромными - формами аутизма.
Еще раз - основное, что нужно знать про CNV в аутизме: они работают в небольшой доле случаев,но их эффект заметен в очень многих аспектах, от дисморфизмов до интеллекта. Т.е. наличие дефектов в CNV почти всегда можно увидеть либо прямо на лице, либо в виде довольно очевидных нарушений типа системной гипотонии и атаксии...
Некоторые известные и документированные CNV

При этом наличие данных CNV не гарантирует аутизма как диагноза. Пенетрантность не 100% никогда. Недавнее эстонское исследование группы людей без каких-либо диагнозов показало, что носители CNV 16p11.2 демонстрируют диcморфизмы головы, ожирение, нарушение когнитивных способностей - но не имеют диагноза аутизма, вопреки данным в таблице выше;
Существует онлайн-база данных CNV, вызывающих аутизм и сходные синдромы http://projects.tcag.ca/autism/
Синдромный аутизм
Синдромным аутизм называют тогда, когда считается, что аутистические симптомы вызваны понятным генетическим расстройством - как правило CNV, но не только (см. Fragile X например). Многие ученые говорят, что термин этот некорректный. Правильнее наверное говорить "аутизм понятной генетической этиологии" или что-нибудь в этом роде.
Очень хороший обзор самых известных форм аутизма с "понятной генетической этиологией" - т.е. синдромных форм аутизма можно найти в блоге Эмили Казанова
- Часть 1: синдромы Тимоти, Smith-Lemli-Opitz, CHARGE, Cornelia de Lange, Lujan-Fryns
Еще раз про пенетрантность
Пенетрантность - penetrance - это термин, означающий процент носителей мутации, у которой проявляется (в данном случае) аутизм. В обзоре в предыдущей главе видно, что пенетрантность для синдромных форм аутизма от 60 до 90 процентов
Пенетрантность в 92% для idic(15) называют "ошеломительной". Да, это много. Это более чем достаточно, чтобы считать данный генетический вариант причиной аутизма - если он найден у ребенка и симптомы сходятся. Это же дает основания полагать, что если эту мутацию исключить при PGD - у следующего ребенка тех же родителей аутизма не будет.
SNP

Теперь давайте доберемся до самых маленьких генетических вариаций, заключенных всего в одном нуклеотиде, одной “букве” В человеческом геноме есть несколько миллионов точек, где распространены вариации в одной “букве” генокода; вокруг все стабильно, а вот в этих конкретных местах - у разных людей по разному. Причем такие вариации почему-то почти всегда биаллельны, то есть там только два варианта “букв” из четырех, и присутствуют они у очень большой доли населения - часто один вариант у 60%, а другой - у 40%. Это не мутация, а полиморфизм, если вспомнить часть 7. (для того, чтобы SNP официально признали таковым, его распространенность должна быть не менее 1%)
То есть это не “мутации”, не ошибки ДНК . Это норма и ее варианты.
Такие вариации называют SNP (читается “снип”), обозначают номерами типа rs2320030 и их найдено и описано порядка 10 миллионов.
Genome-wide association studies
Анализ, показывающий какие варианты (аллели) SNP присутствуют у конкретного человека проводят на SNP чипе. Он представляет из себя пластинку, на которой “напечатаны” образцы генома человека вокруг популярных SNP, ДНК испытуемого (часто образец слюны) наносится на эту пластину и далее смотрят, куда она “прилипла”, а куда нет. Популярный анализ 23andme именно такой, и дает около 1 миллиона таких SNP. Это называется “генотипирование” и оно довольно дешево, а поэтому его применяют сейчас часто (и часто не по делу). Дешево потому, что SNP хорошо известны, и этот анализ удобно делать на потоке, массово.
Эта доступность породила особый вид исследований, Genome-Wide Association Studies (GWAS), при которых берется группа носителей наследуемого признака, и контрольная группа, всех генотипируют, а затем смотрят, нет ли какого-нибудь SNP, который бы у первых был в одном варианте, а у вторых - в другом.
Проблема здесь в статистике: так как SNPов миллионы, велика вероятность того, что мы увидим связь там, где её нет: просто например, на группе в 30 аутистов найдется такой SNP который 30 раз выпадет “правильно” у них. Если монетку бросить миллион раз, она и на ребро встанет:) С этой проблемой можно бороться статистическими же методами, и бороться успешно. Однако в целом GWAS результаты для аутизма отвратительно реплицируются.
Что означает наличие SNP

Эффект от одного или другого варианта SNP как правило мизерный. По другому и быть не может, так как они все по определению очень распространены в популяции. Однако есть заболевания (например муковисцидоз) для которых определяющими являются именно они (но как правило не в одном SNP: обычно наличие “плохого” варианта статистически повышает риск расстройства, скажем, на 2%)
Ни одного SNP, связанного хоть сколь-нибудь значимо с аутизмом, не найдено. Кстати, те, что связаны хоть сколько-нибудь - как правило, сидят не в генах, а в управляющих элементах ДНК - т.е. не в экзоме.
Поэтому все тесты, которые используют SNP генотипирование и рассчитывают значимый “риск аутизма” - шарлатанство скорее всего. Есть не мало сайтов, предлагающих загрузить результаты 23andme и получить “персональные” рецепты лечения аутизма. Самый знаменитый - это “доктор” Yasko, продвигающий рецепты от MTHFR “мутаций”. Шарлатанство. Еще раз, это не мутации. MTHFR - важный ген в фолатном обмене, и он вероятно связан с аутизмом, но не на SNP уровне это искать.
Справедливости ради надо сказать, что комбинация SNP (сотен разных снипов, каждый из которых дает мизерный вклад, но в сумме набирается что-то существенное) кажется способна объяснить шизофрению (в отличие от аутизма) - что пытаются выявить почти те же люди, что делали Human Genome Project, и почти столь же титаническими усилиями. Но там речь идет именно что о сотне снипов.
De novo мутации

“Настоящие” мутации, не живущие постоянно в популяции, мутации, которые появляются у конкретных людей и передаются их детям (или возникают у детей), называются de novo мутациями. Они могут быть как большими участками (CNV), как маленькими вставками и удалениями (indels) так и отдельными “буквами”. Этот пост про последние два вида.
Для их обнаружения тесты на дешевых чипах непригодны, геном надо читать подряд, или, как это называется, секвенировать (sequencing). Можно читать как весь геном вообще - whole genome sequencing (дорого), так и только экзом, только часть, кодирующую белки - exome sequencing, (относительно недорого, $1000 за человека). Соответственно, в клинической практике оно не применяется почти совсем.
Таких de novo мутаций в принципе довольно много - каждый из родителей передает ребенку не менее 100 таких, но только малая часть выпадает на значимые места ДНК. Свежие уважаемые публикации (Iossifov и коллеги 2012) оценивают ключевой вклад de novo мутаций в приблизительно 10% случаев аутизма. Если вы были на конференции в мае, там был доклад Nagwa Meguid о “Новом гене и уникальной форме аутизма” - ровно такой случай, de novo мутации (различные) в гене BCKDK.
Однако и в этой простой истории: вот мутация, вот она затронула ген, важный для работы мозга, вот мы продемонстрировали, что этот ген влияет на аутизм - есть большая загадка. Если, в отличие от Ивана Иосифова исследовать семьи не с одним, а с двумя аутистами, как это сделал Yuen и коллеги в этом году, то оказывается, что они ухитряются брать разные мутации от одних и тех же родителей.
Обобщаем и классифицируем виды “мутаций”
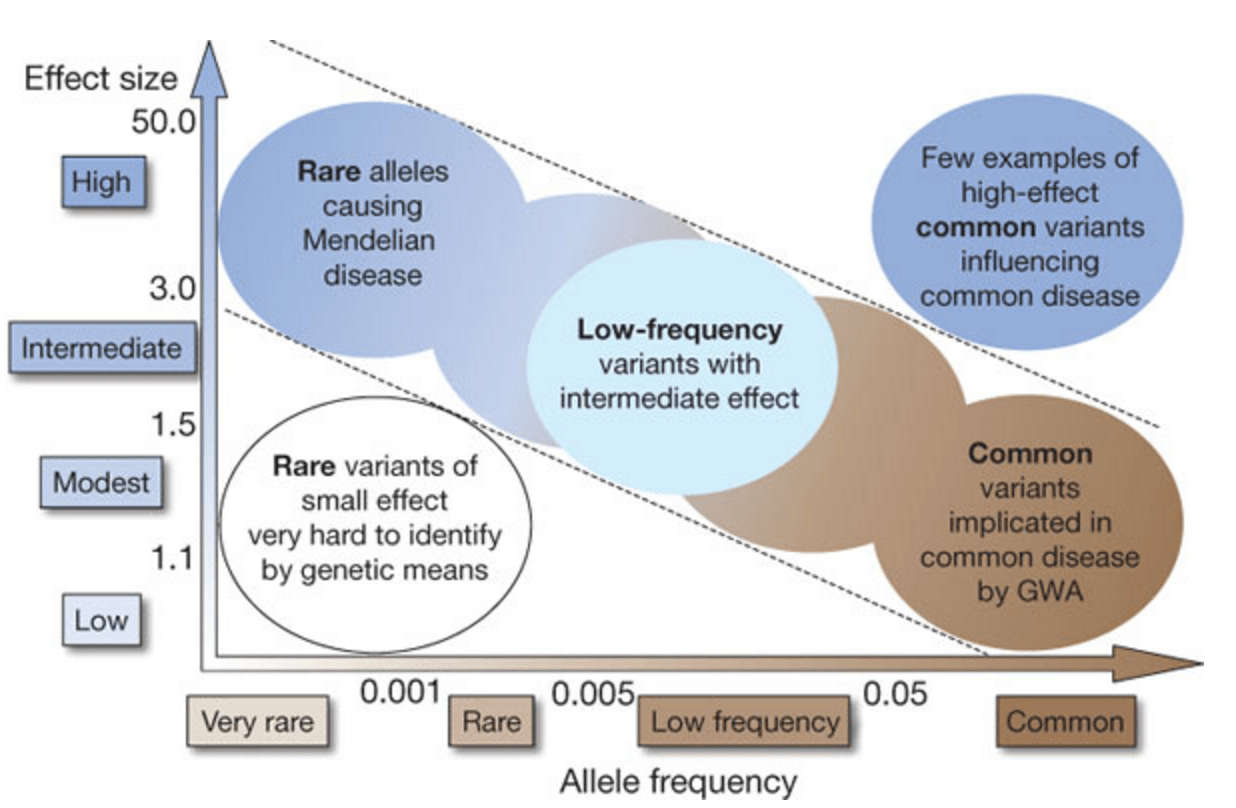
В предыдущих частях мы кратко прошлись по различным видам генетических вариантов в ДНК. От больших по размеру, как правило имеющих заметный эффект, до совсем маленьких.
И правило здесь такое: большие по размеру аномалии как правило редки, но имеют заметный эффект на многие аспекты фенотипа маленькие варианты часто распространены в популяции, но каждая из них имеет мизерный эффект
Это можно суммировать на диаграме типа вот такой, где по горизонтали - распространенность аллели (генетического варианта), от редких к частым, а по вертикали - проявленный эффект у носителей.
Надо отметить, что для аутизма и шизофрении даже самые “сильные” CNV не имеют менеделевского эффекта, обычно повышая риск развития клинически определяемого заболевания с 1% до 3%-10%. Это называется incomplete penetrance . Можно быть носителем известного “шизофренического CNV”, можно иметь дизморфизмы и отклонения в эндокринной системе, характерные для него, но не иметь шизофрении, хотя и обнаруживать некоторые субклинические отклонения.
Соответственно, индивидуальный эффект от полиморфизмов типа SNP вообще мизерный и почти не имеет практического значения для диагностики аутизма.
| Ликбез по генетике аутизма |
|---|
| I. Ликбез по генетике аутизма II. Генетика и близнецы III. Виды мутаций IV. |
Шизофрения - одна из самых загадочных и сложных болезней, причем во многих смыслах. Ее тяжело диагностировать - до сих пор нет консенсуса о том, одно это заболевание или много похожих друг на друга. Ее сложно лечить - сейчас есть лишь препараты, которые подавляют т. н. позитивные симптомы (вроде бреда), но они не помогают возвратить человека к полноценной жизни. Шизофрению сложно исследовать - ни одно другое животное кроме человека ей не болеет, поэтому и моделей для ее изучения почти нет. Шизофрению очень тяжело понять с генетической и эволюционной точки зрения - она полна противоречий, которые биологи пока не могут разрешить. Однако хорошие новости заключаются в том, что в последние годы, наконец, дело вроде бы сдвинулись с мертвой точки. Об истории открытия шизофрении и первых результатах ее изучения нейрофизиологическими методами мы уже . На этот раз речь пойдет о том, как ученые ищут генетические причины возникновения болезни.
Важность этой работы даже не в том, что шизофренией болеет почти каждый сотый человек на планете и прогресс в этой области должен хотя бы радикально упростить диагностику, - даже если создать хорошее лекарство сразу и не получится. Важность генетических исследований в том, что они уже сейчас меняют наши представления о фундаментальных механизмах наследования сложных признаков. Если ученым все-таки удастся понять, как может «прятаться» в нашей ДНК такая сложная болезнь как шизофрения, это будет означать радикальный прорыв в понимании организации генома. И значение такой работы выйдет далеко за пределы клинической психиатрии.
Сначала
немного сырых фактов. Шизофрения
- это тяжелое, хроническое, ведущее к
инвалидности психическое заболевание,
поражающее обычно людей в молодом
возрасте. От нее
страдает около 50 миллионов человек по
всему миру (немногим менее 1% популяции).
Заболевание сопровождается апатией,
безволием, часто галлюцинациями, бредом,
дезорганизацией мышления и речи,
моторными нарушениями. Симптомы обычно
становятся причиной социальной изоляции
и снижения работоспособности. Повышенный
риск суицида у больных шизофренией, а
также сопутствующие соматические
заболевания приводят к тому, что общая
продолжительность жизни у них снижается
на 10-15 лет. Кроме того, больные шизофренией
имеют меньше детей:
мужчины имеют в среднем на 75 процентов, женщины
- на 50 процентов .
Последние полвека стали временем бурного прогресса во многих областях медицины, однако этот прогресс почти не затронул профилактику и лечение шизофрении. Не в последнюю очередь это связано с тем, что мы до сих пор не имеем внятного представления о том, нарушение каких именно биологических процессов является причиной развития заболевания. Такой дефицит понимания привел к тому, что со времени появления на рынке первого антипсихотического препарата хлорпромазина (торговое название: «Аминазин») более 60 лет назад так и не произошло качественного изменения в лечении болезни. Все ныне существующие одобренные для лечения шизофрении антипсихотики (как типичные, включая хлорпромазин, так и атипичные) имеют один и тот же основной механизм действия: они снижают активность дофаминовых рецепторов, что устраняет галлюцинации и бред, но, к сожалению, слабо влияет на негативную симптоматику вроде апатии, безволия, расстройств мышления и т. д. Про побочные эффекты мы даже не упоминаем. Общее разочарование в исследованиях шизофрении проявляется в том, что фармацевтические компании уже давно уменьшают финансирование разработки антипсихотиков , - и это при том, что общее число клинических испытаний только растет. Однако надежда на прояснение причин возникновения шизофрении пришла с довольно неожиданной стороны - она связана с беспрецедентным прогрессом в молекулярной генетике.
Коллективная ответственность
Еще первые исследователи шизофрении заметили, что риск заболеть тесно связан с наличием больных родственников. Попытки установить механизм наследования шизофрении были предприняты почти сразу после переоткрытия законов Менделя, в самом начале XX века. Однако, в отличие от многих других болезней, шизофрения никак не хотела укладывалась в рамки простых менделевских моделей. Несмотря на высокую наследуемость, связать ее с одним или несколькими генами не получалось, поэтому к середине века все большей популярностью стали пользоваться т. н. психогенные теории развития болезни. В согласии с крайне популярным к середине века психоанализом, эти теории объясняли видимую наследуемость шизофрении не генетикой, а особенностями воспитания и нездоровой атмосферой внутри семьи. Появилось даже такое понятие как «шизофреногенные родители».
Однако теория эта, не смотря на свою популярность, прожила недолго. Окончательную точку в вопросе о том, является ли шизофрения наследственной болезнью, поставили психогенетические исследования, проведенные уже в 60-70-е годы. Это были прежде всего близнецовые исследования, а также исследования приемных детей. Суть близнецовых исследований заключается в сравнении вероятностей проявления какого-то признака - в данном случае развития заболевания - у одно- и разнояйцевых близнецов. Поскольку разница в действии среды на близнецов не зависит от того однояйцевые они или разнояйцевые, то различия в этих вероятностях должны происходит главным образом от того, что однояйцевые близнецы генетически идентичны, а разнояйцевые имеют в среднем лишь половину общих вариантов генов.
В случае шизофрении оказалось, что конкордантность однояйцевых близнецов более чем в 3 раза превышает конкордантность разнояйцевых: для первых она составляет приблизительно 50 процентов, а для вторых - менее 15 процентов. Эти слова надо понимать так: если у вас есть страдающий шизофренией однояйцевый брат-близнец, то вы сами заболеете с вероятностью в 50 процентов. Если же вы с братом разнояйцевые близнецы, то и риск заболеть составляет не более 15 процентов. Теоретические расчеты, которые дополнительно учитывают распространенность шизофрении в популяции, дают оценку вклада наследуемости в развитие болезни на уровне 70-80 процентов. Для сравнения, примерно так же наследуется рост и индекс массы тела - признаки, которые всегда считались тесно связанными с генетикой. Кстати, как оказалось позже, столь же высокая наследуемость характерна для трех из четырех остальных основных психических заболеваний: синдрома дефицита внимания и гиперактивности, биполярного расстройства и аутизма.
Результаты близнецовых исследований полностью подтвердились при изучении детей, которые родились у больных шизофренией и были усыновлены в раннем младенчестве здоровыми приемными родителями. Оказалось, что риск заболеть шизофренией у них не снижен по сравнению с детьми, воспитанными своими родителями-шизофрениками, что однозначно указывает на ключевую роль генов в этиологии.
И здесь мы подходим к одной из самых загадочных особенностей шизофрении. Дело в том, что если она так сильно наследуется и при этом очень негативно влияет на приспособленность носителя (напомним, что больные шизофренией оставляют по крайней мере вдвое меньше потомков, чем здоровые люди), то как ей удается сохраняться в популяции по крайней мере на протяжении ? Это противоречие, вокруг которого во многом и происходит главная борьба между разными теориями, получило название «эволюционного парадокса шизофрении»
До недавнего времени ученым было совершенно неясно, какие именно особенности генома больных шизофренией предопределяют развитие болезни. На протяжении десятилетий горячие споры велись даже не о том, какие именно гены изменены у больных шизофренией, а о том, какова общая генетическая «архитектура» болезни.
Имеется ввиду следующее. Геномы отдельных людей очень похожи друг на друга, отличия в среднем составляют менее 0,1 процента нуклеотидов. Некоторые из этих отличительных особенностей генома довольно широко распространены в популяции. Условно считается, что если они встречаются у более чем одного процента людей, их можно называть распространенными вариантами или полиморфизмами. Считается, что такие распространенные варианты появились в геноме человека более 100,000 лет назад, еще до первой эмиграции из Африки предков современных людей, поэтому они присутствуют обычно в большинстве человеческих субпопуляций. Естественно, что для того, чтобы существовать в значительной части популяции на протяжении тысяч поколений большая часть полиморфизмов должна быть не слишком вредна для своих носителей.
Однако в геноме каждого из людей есть и другие генетические особенности,- более молодые и более редкие. Большая часть из них не предоставляет носителям какого-либо преимущества, поэтому их частота в популяции, даже если они фиксируются, остается незначительной. Многие из этих особенностей (или мутаций) имеют более или менее выраженное отрицательное влияение на приспособленность, поэтому они постепенно удаляются негативным отбором. Им взамен в результате непрерывного мутационного процесса появляются другие новые вредные варианты. В сумме частота любой из новых мутаций почти никогда не превышает 0,1 процентов, и такие варианты называют редкими.
Так вот, под архитектурой болезни имеется ввиду то, какие именно генетические варианты - распространенные или редкие, имеющие сильный фенотипический эффект или лишь слегка увеличивающие риск развития болезни, - предопределяют ее появление. Именно вокруг это вопроса до недавнего времени и велись основные споры о генетике шизофрении.
Единственный факт, бесспорно установленный молекулярно-генетическими методами относительно генетики шизофрении за последнюю треть XX века - ее невероятная сложность. Сегодня очевидно, что предрасположенность к болезни определяется изменениями в десятках генов. При этом все предложенные за это время «генетические архитектуры» шизофрении можно объединить в две группы: модель «распространенная болезнь - распространенная изменчивость» («common disease - common variants», CV) и модель «распространенная болезнь - редкие варианты» («common disease - rare variants», RV). Каждая из моделей давала свои объяснения «эволюционного парадокса шизофрении».
RV vs. CV
Согласно модели CV генетическим субстратом шизофрении является некий набор генетических особенностей, полиген, - сродни тому, что определяет наследование количественных признаков вроде роста или массы тела. Такой полиген - это набор полиморфизмов, каждый из которых лишь немного влияет на физиологию (они называются «каузальными», т. к. хоть и не по одиночке, но приводят к развитию болезни). Чтобы поддерживать характерный для шизофрении довольно высокий уровень заболеваемости необходимо, чтобы этот полиген состоял из распространенных вариантов - ведь собрать в одном геноме много редких вариантов очень сложно. Соответственно и каждый человек имеет десятки таких рискованных вариантов в своем геноме. Суммарно все каузальные варианты определяют генетическую предрасположенность (liability) каждого отдельного человека к заболеванию. Предполагается, что для качественных сложных признаков, таких как шизофрения, имеется некое пороговое значение предрасположенности, и заболевание развивается только у тех людей, чья предрасположенность превышает это пороговое значение.
Пороговая модель предрасположенности к заболеванию. Показано нормальное распределение предрасположенности, отложенной по горизонтальной оси. У людей, чья предрасположенность превышает пороговое значение, развивается заболевание.
Впервые такая полигенная модель шизофрении была предложена в 1967 году одним из основателей современной психиатрической генетики Ирвингом Готтесманом, внесшим также значительный вклад в доказательство наследственной природы болезни. С точки зрения приверженцев модели CV сохранение высокой частоты каузальных вариантов шизофрении в популяции на протяжении многих поколений может иметь несколько объяснений. Во-первых, каждый отдельный такой вариант имеет довольно незначительное влияние на фенотип, такие «квази-нейтральные» варианты могут быть невидимы для отбора и оставаться распространенными в популяциях. Особенно это касается популяций с низкой эффективной численностью, где влияние случайности не менее важно, чем давление отбора - к таковым относится и популяция нашего вида.
С другой стороны, выдвигались предположения о присутствии в случае шизофрении т. н. балансирующего отбора, т. е. позитивного влияния «шизофренических полиморфизмов» на здоровых носителей. Это не так уж и сложно представить. Известно, например, что для шизоидных личностей с высокой генетической предрасположенностью к шизофрении (которых много среди близких родственников больных), характерен повышенный уровень творческих способностей, что может слегка увеличивать их адаптацию (это показано уже в нескольких работах). Популяционная генетика допускает такую ситуацию, когда положительный эффект каузальных вариантов у здоровых носителей может перевешивать негативные последствия для тех людей, у которых этих «хороших мутаций» оказалось слишком много, что привело к развитию болезни.
Вторая базовая модель генетической архитектуры шизофрении - модель RV. Она предполагает, что шизофрения - это собирательное понятие и каждый отдельный случай или семья историей заболевания - это отдельная квази-менделевская болезнь, связанная в каждом отдельном случае с уникальными изменениями в геноме. В рамках этой модели каузальные генетические варианты находятся под очень сильным давлением отбора и довольно быстро удаляются из популяции. Но так как в каждом поколении происходит небольшое количество новых мутаций, то между отбором и возникновением каузальных вариантов устанавливается некое равновесие.
С одной стороны, модель RV может объяснить, почему шизофрения очень хорошо наследуется, но ее универсальных генов до сих пор не найдено: ведь в каждой семье наследуются свои собственные каузальные мутации, а универсальных просто нет. С другой стороны, если руководствоваться этой моделью, то приходится признать, что мутации в сотнях разных генов могут приводить к одному и тому же фенотипу. Ведь шизофрения - заболевание распространенное, а возникновение новых мутаций происходит редко. Например, данные по секвенированию троек отец-мать-ребенок показывают, что в каждом поколении на 6 миллиардов нуклеотидов диплоидного генома возникает лишь 70 новых однонуклеотидных замен, из которых в среднем только несколько теоретически могут оказывать какое-либо влияние на фенотип, а мутации других типов - еще более редкое явление.
Тем не менее, некоторые эмпирические данные косвенно подтверждают такую модель генетической архитектуры шизофрении. Например, в начале 90-х годов было обнаружено, что около одного процента всех больных шизофренией имеют микроделецию в одной из областей 22-ой хромосомы. В подавляющем большинстве случаев эта мутация не наследуется от родителей, а происходит de novo в ходе гаметогенеза. Один из 2000 людей рождается с такой микроделецией, приводящей к разнообразным нарушениям в работе организма, названным «синдромом Ди Джорджи». Для страдающих этим синдромом характерны серьезные нарушения когнитивных функций и иммунитета, часто они сопровождаются гипокальциемией, а также проблемами с сердцем и почками. У четверти больных синдромом Ди Джорджи развивается шизофрения. Заманчиво было бы предположить, что и другие случаи шизофрении объясняются сходными генетическими нарушениями с катастрофическими последствиями.
Другим эмпирическим наблюдением косвенно подтверждающим роль de novo мутаций в этиологии шизофрении является связь риска заболеть с возрастом отца. Так, по некоторым данным среди тех, чьим отцам было больше 50 лет на момент рождения, в 3 раза больше больных шизофренией, чем среди тех, чьим отцам было меньше 30. С другой стороны, довольно давно выдвигались гипотезы о связи возраста отца с возникновением de novo мутаций. Такая связь, например, давно установлена для спорадических случаев другой (моногенной) наследственной болезни - ахондроплазии. Эта корреляция совсем недавно была подтверждена вышеупомянутыми данными по секвенированию троек: количество de novo мутаций связано с возрастом отца, но не с возрастом матери. По расчетам ученых от матери ребенок в среднем получает 15 мутаций независимо от ее возраста, а от отца - 25, если ему 20 лет, 55, если ему 35 лет и более 85, если он старше 50. То есть количество de novo мутаций в геноме ребенка увеличивается на две с каждым годом жизни отца.
Казалось, что вместе эти данные довольно ясно указывают на ключевую роль de novo мутаций в этиологии шизофрении. Однако ситуация на самом деле оказалась гораздо сложнее. Уже после разделения двух основных теорий, на протяжении десятилетий генетика шизофрении находилась в стагнации. Не было получено почти никаких достоверных воспроизводимых данных в пользу одной из них. Ни об общей генетической архитектуре болезни, ни о конкретных вариантах, влияющих на риск развития заболевания. Резкий скачок произошел за последние 7 лет и он связан прежде всего с технологическими прорывами.
В поисках генов
Секвенирование первого генома человека, последующее усовершенствование технологий секвенирования, а затем появление и повсеместное внедрение высокопроизводительного секвенирования позволили наконец получить более или менее полное представление о структуре генетической вариабельности в человеческой популяции. Эта новая информация сразу стала использоваться для полномасштабного поиска генетических детерминант предрасположенности к тем или иным заболеваниям, в том числе и к шизофрении.
Строятся подобные исследования примерно так. Сначала собирается выборка неродственных больных людей (cases) и примерно такая же по размеру выборка неродственных здоровых индивидуумов (controls). У всех этих людей определяется наличие тех или иных генетических вариантов - как раз в последние 10 лет у исследователей появилась возможность определять их на уровне целых геномов. Затем производится сравнение частоты встречаемости каждого из определенных вариантов между группами больных людей и группой контроля. Если при этом удается найти статистически достоверное обогащение того или иного варианта у носителей, его называют ассоциацией. Таким образом среди необъятного числа существующих генетических вариантов находятся те, которые связаны с развитием болезни.
Важной величиной, характеризующей эффект ассоциированного с болезнью варианта, является OD (odds ratio, отношение рисков), которое определяется как отношение шансов заболеть у носителей данного варианта по сравнению с теми людьми, у которых он отсутствует. Если величина OD варианта равна 10, это означает следующее. Если взять случайную группу носителей варианта и равную ей группу людей, у которых данный вариант отсутствует, окажется, что в первой группе больных будет в 10 раз больше, чем во второй. При этом чем ближе OD к единице у данного варианта, тем бóльшая выборка нужна для того, чтобы достоверно подтвердить то, ассоциация действительно существует, - что это генетический вариант действительно влияет на развитие болезни.
Подобные работы позволили к настоящему времени обнаружить по всему геному более десятка субмикроскопических делеций и дупликаций , ассоциированных с шизофренией (их называют CNV - copy number variations, одна из CNV как раз вызывает уже известный нам синдром Ди Джорджи). Для обнаруженных CNV, вызывающих шизофрению, OD колеблется в интервале от 4 до 60. Это высокие значения, однако из-за чрезвычайной редкости даже суммарно все они объясняют только очень небольшую часть наследуемости шизофрении в популяции. Что же отвечает за развитие болезни у всех остальных?
После сравнительно неудачных попыток найти такие CNV, которые бы вызывали развитие болезни не в нескольких редких случаях, а у значительной части популяции, сторонники «мутационной» модели возлагали большие надежды на другой тип экспериментов. В них сравнивают у больных шизофренией и здоровых контролей не наличие массивных генетических перестроек, а полные последовательности геномов или экзомов (совокупностей всех кодирующих белки последовательностей). Такие данные, получаемые с использованием высокопроизводительного секвенирования, позволяют находить редкие и уникальные генетические особенности, которые невозможно обнаружить другими методами.
Удешевление секвенирования сделало в последние годы возможным эксперименты такого типа на довольно больших выборках - включающих в последних работах несколько тысяч больных и столько же здоровых контролей. Каков результат? Увы, пока удалось обнаружить лишь один ген, редкие мутации в котором достоверно ассоциированы с шизофренией - это ген SETD1A , кодирующий один из важных белков, участвующих в регуляции транскрипции. Как и в случае с CNV, проблема тут та же самая: мутации в гене SETD1A не могут объяснять сколько-нибудь значимой части наследуемости шизофрении из-за того, что они просто очень редкие.

Связь распространенности ассоциированных генетических вариантов (по горизонтальной оси) и их влияния на риск развития шизофрении (OR). На основном графике красными треугольниками показаны некоторые из обнаруженных к настоящему времени CNV, ассоциированные с болезнью, синими кружками – SNP по данным GWAS. Во врезе в тех же координатах представлены области редких и частых генетических вариантов.
Есть указания на то, что существуют и другие редкие и уникальные варианты, которые влияют на предрасположенность к шизофрении. И дальнейшее увеличение выборок в экспериментах с использованием секвенирования должно помочь отыскать некоторые из них. Однако, несмотря на то, что исследование редких вариантов еще может принести некоторое количество ценной информации (особенно эта информация будет важна для создания клеточных и животных моделей шизофрении), большинство ученых в настоящее время сходятся во мнении, что редкие варианты играют лишь второстепенную роль в наследуемости шизофрении, а модель CV намного лучше описывает генетическую архитектуру болезни. Убежденность в верности CV модели пришла прежде всего с развитием исследований типа GWAS, о которых мы подробно расскажем во второй части. Коротко говоря, исследования такого типа позволили обнаружить ту самую распространенную генетическую изменчивость, описывающую значительную долю наследуемости шизофрении, существование которой предсказывалось моделью CV.
Дополнительным подтверждением CV модели для шизофрении является связь между уровнем генетической предрасположенности к шизофрении и так называемыми расстройствами шизофренического спектра. Еще ранние исследователи шизофрении заметили, что среди родственников больных шизофренией часто встречаются не только другие больные шизофренией, но и «эксцентрические» личности со странностями характера и симптоматикой сходной с шизофренической, но выраженной менее ярко. Впоследствии подобные наблюдения привели к концепции, согласно которой существует целый набор болезней, для которых характерны более или менее выраженные нарушения в восприятии реальности. Эта группа болезней получила название расстройства шизофренического спектра. Помимо различных форм шизофрении к ним относят бредовые расстройства, шизотипическое, параноидное и шизоидное расстройства личности, шизоаффективное расстройство и некоторые другие патологии. Готтесман, предлагая свою полигенную модель шизофрении, предположил, что у людей с субпороговыми значениями предрасположенности к болезни могут развиваться другие патологии шизофренического спектра, причем тяжесть заболевания коррелирует с уровнем предрасположенности.
Если эта гипотеза верна, логично предположить, что генетические варианты, обнаруженные как ассоциированные с шизофренией, будут обогащены и среди людей, страдающих расстройствами шизофренического спектра. Для оценки генетической предрасположенности каждого отдельного человека используется специальная величина, называемая уровнем полигенного риска (polygenic risk score). Уровень полигенного риска учитывает суммарные вклад всех идентифицированных в GWAS распространенных рискованных вариантов, имеющихся в геноме данного человека, в предрасположенность к болезни. Оказалось, что, как и предсказывала модель CV, значения уровня полигенного риска коррелируют не только с самой шизофренией (что тривиально), но и с другими болезнями шизофренического спектра, причем тяжелым типам расстройств соответствуют более высокие уровни полигенного риска.
И все-таки остается одна проблема - феномен «старых отцов». Если большая часть эмпирических данных подтверждает полигенную модель шизофрении, как согласовать с ней давно известную связь между возрастом отцовства и риском детей заболеть шизофренией?
Некогда было выдвинуто изящное объяснение этого феномена с точки зрения модели CV. Предполагалось, что позднее отцовство и шизофрения не являются соответственно причиной и следствием, а представляют собой два следствия общей причины, а именно генетической предрасположенности поздних отцов к шизофрении. С одной стороны, высокий уровень предрасположенности к шизофрении может коррелировать у здоровых мужчин с более поздним отцовством. С другой стороны, очевидно, что высокая предрасположенность отца предопределяет повышенную вероятность того, что его дети заболеют шизофренией. Выходит, что мы можем иметь дело с двум независимыми корелляциями, а значит накопление мутаций в предшественниках сперматозоидов у мужчин можетпочти никак не влиять на развитие шизофрении у их потомков. Недавно полученные результаты моделирования , учитывающего эпидемиологические данные, а также свежие молекулярные данные по частоте de novo мутаций, хорошо согласуются именно с таким объяснением феномена «старых отцов».
Таким образом, в настоящий момент можно считать, что убедительных аргументов в пользу «мутационной» RV модели шизофрении уже почти не осталось. А значит ключ к этиологии болезни лежит в том, какой именно набор распространенных полиморфизмов вызывает шизофрению в соответсвии с CV-моделью. Тому, как этот набор ищут генетики и что им уже удалось обнаружить, будет посвящена вторая часть нашей истории.
Аркадий ГоловНейрогенетика и генетика наследственных заболеваний
Диагностика основных микродупликационных и микроделеционных синдромов (код теста 01.02.05.300)
Синдром 1р36 микроделеции вызван делецией (в 7% случаев - транслокацией) участка короткого плеча (р) 1-й хромосомы (1р-моносомия). От конкретного региона и вида делеции (терминальная, интерстициальная, комплексные перестройки) зависит выраженность симптомов. Клинически проявляется отставанием в развитии, мышечным гипотонусом, черепно-лицевыми аномалиями: прямые брови, глубоко посаженные глаза, ретрузия средней части лица, широкая и вогнутая переносица, удлиненный фильтрум, заостренный подбородок, большой, длительно заживающий родничок, микробрахицефалия, эпикантус, повернутые кзади низко посаженные ушные раковины, брахи- и камптодактилия и укороченные нижние конечности, возможны судорожные припадки. К другим особенностям относятся структурные аномалии мозга, врожденные пороки сердца, зрительные и глазные нарушения, тугоухость, аномалии развития скелета, наружных половых органов и почек.
Чаще всего мутация возникает de novo, но в редких случаях может появляться при наличии у одного из родителей сбалансированной (скрытой) перестройки - транслокации, затрагивающей регион 1p36. Носители сбалансированной транслокации не имеют симптомов болезни, но существует риск 50% передачи мутации последующему поколению. Поэтому, рекомендовано проведение молекулярно-генетического обследования родителей пациента с подтвержденным 1р36 микроделеционным синдромом.
Исследование генов:
- TNFRSF4
GNB1
GABRD
Синдром 2p16.1-p15 микроделеции вызван делецией 16.1-15 участков короткого плеча (р) 2-й хромосомы. Делеция участка хромосомы может захватывать до 12 известных генов. Клинически признаки включают задержку психомоторного и речевого развития и черепно-лицевые аномалии, такие как: телекантус, опущение век и наружных уголков глаз, узкую глазную щель (антимонголоидный разрез глаз), выдающуюся переносицу, высокое небо, удлиненный фильтрум, вывернутую верхнюю губу. У некоторых пациентов имеются микроцефалия, гипоплазия зрительного нерва, аномалии развития почек и гидронефроз, увеличенный размер сосков, низкий рост, кортикальная дисплазия, камптодактилия и деформация пальцев ног в виде так называемой «голубиной стопы».
Во всех описанных случаях делеция возникла de novo и риск наследования данного заболевания сибсами равен среднему популяционному значению. В случае наличия у родителей сбалансированной транслокации или герминативного мозаицизма риск возникновения заболевания у сибсов выше относительно среднего популяционного риска, в связи с чем для родителей ребенка с 2p16.1-p15 микроделеционным синдромом рекомендован молекулярно-генетический анализ.
Исследование генов:
REL
PEX13
Синдром 2q23.1 микроделеций/микродупликаций вызывается утратой (делецией) или удвоением (дупликацией) участка длинного плеча (q) 2-й хромосомы в положении 23.1, в критическом регионе которого расположен ген MBD5 или некоторые из его экзонов (интерстициальные делеции ~5% случаев). Также возможен гетерозиготный вариант патогенной последовательности гена MBD5 (~5%). Данный ген является дозо-чувствительным, поэтому уменьшение (делеция) или увеличение (дупликация) дозы гена приводят к развитию 2q23.1 микроделеционного/микродупликационного синдрома.
Данное заболевание характеризуется общим отставанием в развитии, тяжелыми нарушениями речи (большинство пациентов не способны говорить или говорят отдельные слова, короткие фразы или предложения), припадками, дебют которых приходится на возраст двух лет; нарушениями сна, проявляющимися в виде чрезмерной дневной сонливости, и девиантным поведением, включающим в себя аутистическое поведение, намеренное нанесение себе телесных повреждений и агрессивное поведение. К другим клиническим признакам относятся микроцефалия, широкий рот,вздернутая верхняя губа, выдающиеся резцы, опущенные уголки рта, макроглоссия, аномалии развития уха.
Делеция и дупликация возникают de novo, однако были описаны случаи наследования заболевания от родителя по аутосомно-доминантному типу, что может быть связано со сниженной пенетрантностью. В связи с этим генетическая диагностика рекомендована обоим родителям для вычисления риска заболевания у сибсов.
Исследование генов:
MBD5
Делеция 2q23.1, содержащая ген MBD5 или его часть (~90% пациентов)
Интерстициальная делеция, содержащая один и более экзонов гена MBD5 (~5%)
Гетерозиготный вариант патогенной последовательности гена MBD5 (~5%)
SATB2 - ассоциированный синдром вызван нарушениями в работе гена SATB2, локализованного в длинном плече (q) 2-й хромосомы в положении 32-33, вследствие делеции, дупликации, транслокации или точеченых мутаций. Ген SATB2 кодирует одноименный белок, участвующий в нормальном развитии нервной и костной систем, в том числе лицевых структур. К основным симптомам относят тяжелые нарушения речи, аномалии развития неба, костей и мозга, поведенческие нарушения. Дебют заболевания приходится на возраст 2-х лет.
Мутация возникает de novo и наследуется по аутосомно-доминантному типу. В случае наличия у родителей сбалансированной транслокации или герминативного мозаицизма риск возникновения заболевания у сибсов выше относительно среднего популяционного риска, в связи с чем для родителей ребенка с SATB2- ассоциированным синдромом рекомендован молекулярно-генетический анализ.
Исследование генов:
- SATB2
Крупные делеции, интрагенные делеции и дупликации, и перестройки, включающие SATB2, точечные мутации.
Синдром 3q29 микроделеции/микродупликации вызван делецией или дупликацией 29-го участка длинного плеча (q) 3-й хромосомы. Для пациентов с микродупликацией характерно отставание в развитии, микроцефалия и офтальмологические нарушения, аномалии развития сердца; мышечный гипотонус, задержка речевого развития, краниосиностоз, высокое «готическое» небо, зубочелюстные аномалии, кондуктивная тугоухость, аномалии опорно-двигательного аппарата; припадки. Часто у многих носителей данной дупликации не наблюдается выраженной симптоматики, что связано со сниженной пенетрантностью.
Мутация может возникать de novo или может наследоваться от родителя в отсутствие клинической симптоматики, имеющего данную перестройку.
3q29 микроделеционный синдром клинически проявляется отставанием в ключевых этапах развития ребенка (сидение, ходьба, речь), частыми отитами и респираторными инфекциями, микроцефалией. Некоторые дети рождаются с расщелиной губы или неба, возможно наличие пороков сердца. С возрастом возможно развитие поведенческих и психических нарушений. Клиническая картина крайне вариабельна и некоторые люди с 3q29 делецией могут иметь невыраженную симптоматику или вообще не знать о наличии заболевания.
Мутация возникает de novo, однако при наличии у родителей заболевания в невыраженной степени передача мутации происходит по аутосомно-доминантному типу.
Исследование генов:
- DLG1, но пенетрантность не является 100-процентной.
Синдром Вольфа-Хиршхорна возникает вследствие делеции или несбалансированной транслокации теломерного участка короткого плеча (p) 4-й хромосомы в положении 16 (4p16). Редко у пациентов выявляется так называемая «кольцевая 4-я хромосома», что может произойти в том случае, если делеция возникла на обоих концах хромосомы и последние подверглись слиянию и сформировали кольцевую структуру. Размер делеции может различаться, с чем, вероятно, связана выраженность симптоматики.
Заболевание характеризуется типичными черепно-лицевыми аномалиями, в том числе аномалией развития черепа в виде так называемого “шлема греческого воина” (широкая переносица, сливающаяся с лобной частью черепа), микроцефалией, высокой передней линией роста волос с выдающейся глабеллой, широко посаженными глазами (гипертелоризм), эпикантусом, приподнятыми дугообразными бровями, укороченным фильтрумом, опущенными уголками рта, микрогнатией (недоразвитием верхней челюсти), недостаточным развитием ушных раковин или формированием преаурикулярных выростов. У всех больных отмечается пренатальный дефицит роста, за которым следуют задержка постнатального развития и гипотонус мышц в сочетании с их недоразвитием. Также наблюдается отставание в общем развитии различной степени выраженности, судорожные припадки. К другим симптомам относят аномалии развития скелета, врожденные пороки сердца, глухота (в большинстве случаев кондуктивная, аномалии развития урогенитального тракта, структурные аномалии мозга).
В 85-90% случаях мутация возникает de novo в гаметах или на ранних стадиях развития. В остальных случаях родители являются носителями сбалансированной транслокации, что приводит к формированию несбалансированной транслокации у потомков, включающей в себя как делецию участка 4-й хромосомы (моносомия).
Риск заболевания у сибсов зависит от того, возникла ли делеция de novo (риск заболевания равен среднему популяционному риску) или в результате несбалансированной транслокации (риск заболевания выше среднего популяционного).
Исследование генов:
LETM1
WHSC1(NSD2)
Cиндром кошачьего крика вызван делецией короткого плеча (p) 5-й хромосомы. К основным клиническим проявлениям относятся высокочастотный монотонный плач, микроцефалия, широкая переносица, эпикантус, микрогнатия, измененная дерматоглифика, а также тяжелые психомотрные нарушения и умственная отсталость. Редко встречаются аномалии развития сердца, почек, возможно наличие преаурикулярных выростов, синдактилии, гипоспадии и крипторхизма. Клиническая симптоматика зависит от размера делеции и может сильно варьировать.
В большинстве случаев делеция возникает de novo, то есть вероятность развития заболевания у сибсов равно среднему популяционному риску. Однако в 10% случаев данное состояние наследуется от родителя, несущего сбалансированную перестройку, что ведет к формированию несбалансированной перестройки с делецией у потомка. Для определения вероятности развития болезни у сибсов рекомендовано молекулярно-генетическое обследование обоих родителей.
Для выявления данной мутации используются пробы к генам TERT и SEMA5A. Чувствительность диагностических тестов составляет 90-95%, что связано с невозможностью выявления интерстициальных делеций.
Исследование генов:
- TERT
SEMA5A
Синдром Сотоса вызван делецией участка длинного плеча 5-й хромосомы (5q35) или гетерозиготной мутацией в гене NSD1.
Синдром Сотоса характеризуется тремя важнейшими клиническими проявлениями: специфический внешним вид (широкий выдающийся лоб, редкий волосяной покров во фронто-темпоральной части головы, антимонголоидный разрез глаз, румянец, вытянутое заостренное лицо, острый подбородок), избыточный рост (рост и/или окружность головы более чем в два раза превышает норму), трудности в обучении (раннее отставание в развитии, умственная неполноценность средней и тяжелой степени выраженности). К другим симптомам относятся поведенческие нарушения, раннее окостенение, пороки сердца, аномалии черепа и почек, повышенная гибкость суставов, плоскостопие, сколиоз, неонатальная желтуха, гипотонус мышц, припадки.
Чаще всего мутация возникает de novo при формировании гамет. Обычно при этом у пациентов отсутствует семейный анамнез по данному заболеванию.
В 5% случаев родитель пробанда является носителем патогенной мутации, и, поскольку, наследование заболевание происходит по аутосомно-доминантному типу, риск развития синдрома Сотоса у сибсов равен 50%. Рекомендуется молекулярно-генетическое обследование родителей.
Исследование генов:
- NSD1
Синдроме Вильямса-Бойрена (7q11.23 дупликационный синдром) возникает вследствие дупликации участка длинного плеча 7-й хромосомы. Данный регион является критическим и включает в себя 26-28 генов, а частности, ген ELN, дупликация которого, вероятно, ассоциирована с дилатацией аорты, возникающей при данном синдроме. Помимо этого, заболевание характеризуется повреждениями со стороны сердечно-сосудистой системы (эластиновая артериопатия, периферический стеноз легочной артерии, надклапанный стеноз аорты, гипертензия), характерным внешним видом, соеденительнотканной дисплазией, неврологическими нарушениями (гипотонус мышц, непроизвольные движения, нарушения походки и позы), нарушениями речи (детская апраксия речи, дизартрия, фонологические нарушения), поведенческими нарушениями (тревожное расстройство, агрессивное поведение, селективный мутизм, синдром дефицита внимания и гиперактивности, расстройства аутистического спектра), умственной отсталостью, эндокринными нарушениями (гиперкальциемией, гиперкальциурией, гипотиреозом, ранним половым созреванием). Примерно у 30 % пациентов выявляется один и более пороков развития. Нарушения в питании часто приводят к недостаточному набору веса в младенчестве. Вследствие мышечного гипотонуса и избыточной растяжимости суставов нормальные этапы развития ребенка могут запаздывать.
Дупликация возникает de novo и чаще всего это происходит при формировании гамет. Обычно при этом у пациентов отсутствует семейный анамнез по данному заболеванию. В четверти случаев ребенок наследует хромосому с дуплицированным участком от родителя, имеющего стертую симптоматику. Наследование заболевания происходит по аутосомно-доминантному типу. Риск передачи заболевания потомкам от родителя, несущего хромосому с дупликацией, составляет 50%. Рекомендуется молекулярно-генетический анализ на наличие дупликации у родителей.
Исследование генов:
- ELN
Синдром Лангера-Гидиона (Трихоринофалангеальный синдром II типа) (0.2-1:100,000)
Синдром Лангера-Гидиона (Трихоринофалангеальный синдром II типа) вызван делецией участка 24.11-24.13 длинного плеча 8-й хромосомы, от размера которой зависит выраженность клинических проявлений. Заболевание характеризуется особенностями развития эктодермы (мелкие редкие депигментированные и медленно растущие волосы, ониходистрофия, микромастия), а также деформацией скелета (невысокий рост, укорочение стоп, брахидактилия с ульнарной или радиальной девиацией пальцев кисти, ранние проявления дисплазии тазобедренного сустава), множественными остеохондромами (первоначально они обнаруживаются в области лопатки и около локтевого и коленного суставов в возрасте от 1 месяца до 6 лет) и высоким риском умственной отсталости легкой и средней степени выраженности.
Делеция возникает de novo и чаще всего это происходит при формировании гамет. Обычно при этом у пациентов отсутствует семейный анамнез по данному заболеванию. В некоторых случаях ребенок наследует хромосому с делетированным участком от родителя, имеющего стертую симптоматику. Наследование заболевания происходит по аутосомно-доминантному типу. Риск передачи заболевания потомкам от родителя, несущего хромосому с дупликацией, составляет 50%. Рекомендуется молекулярно- генетический анализ на наличие делеции у родителей.
Исследование генов:
- TRPS1
EXT1
Синдром 9 q 22.3 микроделеции вызван делецией участка 22.3 длинного плеча 9-й хромосомы. Данный участок включает в себя ген PTCH1, мутация в котором приводит к развитию синдрома Горлина (синдром невоидной базальноклеточной карциномы), поэтому клинические проявления этих заболеваний сходны. Также возможны отставание в развитии и/или умственная неполноценность, метопический краниосиностоз, обструктивная гидроцефалия, пре- и постнатальная макросомия, припадки. У больных с микроделеционным синдромом 9q22.3 имеется высокий риск появления опухоли Вильмса (нефробластома). К типичным проявлениям синдрома Горлина относятся: обызвествление серпа головного мозга в возрасте до 20 лет, базальноклеточная карцинома, одонтогенные кератокисты, точечные углубления на ладонях и подошвах; у больных с данным синдромом повышен риск медуллобластомы, а также фибромы сердца и яичников. Симптомы 9q22.3 микроделеционного синдрома крайне вариабельны и зависят от размера микроделеции, который может достигать 270 генов.
Данная мутация может передаваться по наследству (в этом случае родители являются носителем сбалансированной (скрытой) перестройки - транслокации, затрагивающей 9q22.3) или возникать de novo. В случае наличия у родителей сбалансированной транслокации риск возникновения заболевания у сибсов выше относительно среднего популяционного риска, в связи с чем для родителей ребенка с 9q22.3 микроделеционным сидромом рекомендован молекулярно-генетический анализ.
Заболевание передается по аутосомно-доминантному типу и риск передачи мутации от родителя, несущего делецию 9q22.3, потомкам составляет 50%.
Исследование генов:
- FANCC
PTCH1
Синдром ДиДжорджи / Велокардиофациальный синдром возникает вследствие делеции региона 11.2 длинного плеча 22-й хромосомы либо региона 14 короткого плеча 10 хромосомы. Клинически характеризуется врожденными пороками сердца, (тетрада Фалло, атрезия дуги аорты, дефект межжелудочковой перегородки, общий артериальный ствол); дефектами неба (в частности, велофарингеальная недостаточность, врожденная расщелина нёба и одна из ее форм - подслизистая (скрытая) расщелина нёба, расщепленный язычок (uvula)) и характерными чертами лица (данный признак присутствует у большинства пациентов из северной Европы). Помимо этого имеется аплазия тимуса, ведущая к иммуннодефициту, и паращитовидных желез, ведущая к гипокальциемии, а также нарушения кормления и глотания, констипация (в некоторых случаях может сочетаться с аномалиями развития желудочно-кишечного тракта, такими как мальротация, атрезия ануса, болезнь Гиршпрунга), аномалии развития почек, тугоухость (кондуктивная и нейросенсорная), ларинготрахеоэзофагеальные аномалии, недостаток гормона роста (соматотропного гормона), аутоиммунные заболевания, судорожные припадки (идиопатические или ассоциированные с гипокальциемией), аномалии развития ЦНС (синдром фиксированного спинного мозга) и скелета (сколиоз, косолапость, полидактилия, краниосиностоз), офтальмологические нарушения (страбизм, задний эмбриотоксон, ангиопатия сосудов сетчатки, склерокорнеа, анофтальм), гипоплазия эмали, злокачественные заболевания (редко).
Типичны отставание в развитии (в частности, задержка речевого развития), умственная неполноценность, трудности в обучении (однако существует значительное преобладание невербального интеллекта над вербальным). Аутизм и расстройства аутистического спектра встречаются у 20% пациентов детского возраста, психические заболевания (особенно шизофрения) - у 25% взрослых. Частыми являются синдром дефицита внимания, тревожное расстройство, персеверация, нарушение социализации.
В 90% случаев делеция возникает de novo и чаще всего это происходит при формировании гамет. Обычно при этом у пациентов отсутствует семейный анамнез по данному заболеванию. В 10% случаев ребенок наследует хромосому с делетицованным участком от родителя, у которого клинически заболевание может оставаться невыраженным. Наследование заболевания происходит по аутосомно-доминантному типу. Риск передачи заболевания потомкам от родителя, несущего хромосому с делецией, составляет 50%. Рекомендуется молекулярно-генетический анализ на наличие делеции у родителей.
Исследование генов:
- CLDN5, регион AB
GP1BB, регион AB
SNAP29, регион CD
PPIL2; дистальная часть 22q11
RTDR1; дистальная часть 22q11
GATA3
При синдроме Прадера-Вилли и синдроме Ангельмана повреждается один и тот же локус длинного плеча 15-й хромосомы (15q11.2-13), однако клинические проявления этих заболеваний существенно различаются, что связано с разнообразием механизмов их возникновения и причастностью явления геномного импринтинга в их развитии (явление, при котором активность различных генов варьируется в зависимости от их родительского происхождения). Следует отметить, что гены, подвергшиеся мутации при данных заболеваниях (критический регион Прадера-Вилли), в норме «работают» только на отцовской (гены SNRPN) или материнской хромосоме (ген UBEA3), в то время как на материнской или отцовской хромосоме они метилированы и, соответственно, деактивированы.
Имеется несколько причин возникновения синдрома Прадера-Вилли: делеция участка 15-й хромосомы, унаследованной от отца (70% случаев), унипарентеральная (однородительская) дисомия, при которой обе 15-е хромосомы имеют материнское происхождение (соответственно, обе копии генетического материала метилированы и не экспрессируются) (28% случаев). Меньше чем в 1% случаев заболевание возникает вследствие мутации в центре импринтинга отцовской хромосоме. Также возможна несбалансированная транслокация данного региона и эпимутация, вызванная невозможностью деметилирования материнской хромосомы отца при сперматогенезе.
Причинами развития синдрома Ангельмана являются: делеция региона Прадера-Вилли/Ангельмана, локализованного на 15-й хромосоме, унаследованной от матери; мутация гена UBEA3, локализованного на 15-й хромосоме, унаследованной от матери (данный ген импринтирован на отцовской хромосоме), отцовская унипарентеральная дисомия или дефект импринтинга.
Синдром Прадера-Вилли характеризуется мышечным гипотонусом, нарушением кормления в период младенчества, склонностью к перееданию в период раннего детства и постепенным развитием морбидного ожирения. Имеется отставание нормальных этапов речевого и моторного развития. В той или иной степени у всех пациентов имеются когнитивные расстройства. Специфичен поведенческий фенотип, проявляющийся в виде истерик (tempertantrum), упрямства, манипулятивного поведения, обсессивно-компульсивных растройств. Для пациентов обоих полов характерен гипогонадизм, проявляющийся в виде гипоплазии половых органов, неполноценности полового созревания, а также бесплодие. При отсутствии лечения соматотропным гормоном характерен невысокий рост. К другим внешним проявлениям относятся страбизм, сколиоз.
Синдром Ангельмана характеризуется тяжелым отставанием в развитии и умственной неполноценностью, нарушением речи, атактической походкой и/или тремором конечностей, а также уникальным поведенческим паттерном (частый смех, улыбка, возбудимость), который выявляется не раньше первого года жизни. Отставание в развитии обычно обнаруживается в первые полгода жизни. Зачастую правильный диагноз удается поставить только через несколько лет. Также характерны микроцефалия и припадки.
Риск развития синдрома Прадера-Вилли у сибсов различен и зависит от механизма развития генетической перестройки: при интерстициальной делеции, материнской унипарентеральной дисомии и эпимутации риск составляет <1%; при несбалансированной транслокации или интерстициальной делецией в центре импринтинга он может достигать 50%, а при материнской унипарентеральной дисомии с транслокацией-100%. В связи с этим рекомендовано проведение молеулярно-генетического тестирования у родителей.
При синдроме Ангельмана хромосомные перестройки чаще всего возникают de novo при гаметогенезе. Риск развития заболевания у сибсов зависит от причины, вызвавшей мутацию у пробанда: в случае делеции, отцовской унипарентеральной дисомии, дефекта импринтинга риск составляет <1%; при несбалансированной транслокации, интерстициальной делеции центра импринтинга, мутации в гене UBEA3 риск может достигать 50%; при отцовской унипарентеральной дисомии с транслокацией риск достигает 100%.
Исследование генов:
- SNRPN
UBE3A
Синдром дупликации 15q вызван дупликацией участка 15q11.2-q13.1 (так называемого критического региона Прадера-Вилли/Ангельмана), локализованного в длинном плече 15-й хромосомы. В 80% случаев имеются 4 копии критичного региона (тетрасомия 15q11.2-q13.1или idic(15)), в остальных случаях происходит интерситициальная дупликация, при которой имеется 3 копии критического региона (трисомия 15q11.2-q13.1). Обычно выраженность симптоматики снижена у пациентов с трисомией.
Синдром проявляется отставаниями в языковом развитии и моторных навыках, таких как ходьба и сидение, гипотонией, припадками, низкорослостью. Отличительными признаками являются очень тонкие черты лица, однако могу присутствовать такие признаки, как эпикантальные складки (кожные складки во внутренних углах одного или обоих глаз), широкий лоб, сплющенный мост носа, нос «кнопкой», высокое арочное небо. У многих пациентов наблюдаются проявления заболеваний аутистического спектра, такие как нарушения коммуникации и социальных взаимодействий, навязчивые интересы, нарушенные циклы сна (и сниженная потребность во сне) и повторяющиеся и стереотипное поведение. Также, часто наблюдается высокий болевой порог. Если речь развита, то обычно наблюдается эхолалия. У пациентов может отсутствовать способность к ходьбе или речи.
Все известные случаи тетрасомии возникали de novo. При трисомии 85% случаев возникают de novo, а в 15% мутация наследуется по аутосомно-доминантному типу (если мутация представлена интерстициальной делецией) и риск возникновения заболевания у сибсов составляет 50%. В связи с этим рекомендуется генетическое обследование родителей.
Генетические маркеры:
- SNRPN
UBE3A
Синдром делеции 15q24 (синдром Виттевеена-Колька) (3:10,000-4:10,000) характеризуется глобальной задержкой развития, от лёгкой до тяжёлой умственной отсталостью, лицевыми дисморфизмами: высокая линия роста волос, глубоко посаженные глаза, треугольная форма лица. Кроме этого могут наблюдаться врожденные пороки развития кистей и стоп, глаз, половых органов, нестабильность суставов, отставание в росте. Менее распространённые черты - эпилептические приступы, кондуктивная и сенсоневральная тугоухость, гипоспадия и/ или микропения.
Генетические маркеры:
- SEMA7A
CYP1A1
Синдром Рубинштейна-Тейби вызван мутацией или делецией участка короткого плеча 16-й хромосомы, содержащего ген CREBBP, регулирующего рост клеток и их деление и необходимого для нормального развития плода. В 3-8% случаев заболевание вызвано мутацией в гене EP300.
Характеризуется отличительными чертами лица у пациентов (арочные брови, наклоненные вниз пальпебральные щели, низко расположенная перегородка преддверия носа, гримасничающая улыбка, высокое небо,), широкими и часто угловатыми пальцами на руках и большими пальцами на ногах, низким ростом и наличием отставания умственного развития (от умеренной до тяжелой степени). Пренатальное развитие обычно нормальное, тем не менее, центильные показатели роста, веса и окружности головы быстро уменьшаются в первые месяцы жизни. Ожирение может появиться в детстве или подростковом возрасте. Значения IQ колеблются в пределах 25-79 баллов. Другими встречающимися проявлениями могут быть: колобома, катаракта, врожденные пороки сердца, патологии почек и крипторхизм.
Мутация или делеция при данном заболевании возникает de novo. Однако в связи с наличием случаев передачи заболевания по аутосомно-доминантному типу от родителейс невыраженной симптоматикой (связано с соматическим мозаицизмом) и несущих мутацию в гене CREBBP (миссенс мутация, делеция), рекомендуется генетическое обследование родителей. Риск развития заболевания у сибсов в данном случае составляет 50%.
Генетические маркеры:
CREBBP
LIS 1-ассоциированная лиссэнцефалия (синдром Миллера-Дикера)/ изолированная лиссэнцефалия/ синдром двойной коры (от 11.7 до 40 на миллион рождений). Лиссэнцефалия и синдром двойной коры представляют собой кортикальные мальформации, вызванные недостаточной миграцией нейронов во время эмбриогенеза. Лиссэнцефалияхарактеризуется нарушением развития извилин головного мозга -агирия и пахигирия. Синдром двойной коры относится к группе гетеротопий серого вещества. При данной нозологии серое вещество локализованно прямо под корой головного мозга и отделенно от него тонкой зоной нормального белого вещества. Синдром Миллера-Дикера характеризуется лиссэнцефалией, аномалиями черепно-лицевого скелета и серьезными неврологическими аномалиями. Изолированная лиссэнцефалия характеризуется лиссэнцефалией и ее прямыми последствиями: отставанием в развитии, умственной ндостаточностью и припадками.
Генетические маркеры:
PAFAH1B1 (LIS1)
Синдром Смита-Магениса (синдром делеции 17 p 11.2) (1:15,000) характеризуется черепно-лицевыми аномалиями, прогрессирующими с возрастом, отставанием в развитии, когнитивными нарушения и аномалиями поведения. У младенцев возникают проблемы с кормлением, задержка роста, гипотония, гипорефлексия, длительная дремота и необходимость будить младенцев для кормления и генерализованная летаргия. Большинство пациентов имеют отставание умственного развития. Поведенческий паттерн, включающий значительное нарушение сна, стереотипию и аутотравматичное поведение, обычно не выявляется до 18 месяцев. Нарушения поведения обычно проявляются невнимательностью, отвлекаемостью, гиперактивностью, импульсивностью, частыми вспышками гнева, поиском внимания, непослушанием, агрессией, трудностями с туалетом и самоповреждающим поведением.
У людей с синдромом дупликации 17p11.2 (синдром Потоцки-Лупски) часто наблюдается гипотония, недостаточное питание и снижения скорости развития в младенчестве. Они также страдают нарушениями в развитии моторных и умственных способностей. Кроме того, поведенческие паттерны у многих пациентов представлены спектром аутических нарушений. В большинстве случаев, синдром Потоцки-Лупски развивается спорадически, но иногда может наследоваться.
Генетические маркеры:
RAI1
DRC3
LLGL1
Нейрофиброматоз 1 типа , вызванного делецией гена NF1, возникает вследствие делеции участка длинного плеча 17-й хромосомы (17q11.2), содержащего ген NF1, кодирующий белок нейрофибромин, содержащийся в олигодендроцитах и подавляющий опухолевую активность.
Клинически характеризуется множественными пятнами на коже цвета кофе с молоком, пигментными пятнами в подмышечных и паховой областях, множественными кожными нейрофибромами и узелками Лиша на радужной оболочке. Трудности в обучении имеют место как минимум у 50% больных с нейрофиброматозом 1 типа. Менее распространёнными проявлениями являются плексиформные нейрофибромы, глиомы зрительного нерва и других участков ЦНС, злокачественные опухоли оболочек периферических нервов, сколиоз, тибиальная дисплазия и васкулопатия. У больных с делецией гена NF1 часто присутствует более тяжёлый фенотип заболевания.
Заболевание наследуется по аутосомно-доминантному типу. Имеется риск 50% передачи мутационной аллеи последующему поколению.
Генетические маркеры:
NF1
Синдром KANSL 1-связанной умственной отсталости (1: 16,000) характеризуется неонатальной/детской гипотонией, дисморфизмом, врожденными пороками и характерными поведенческими проявлениями. У всех пациентов с раннего детства отмечается психомоторное отставание в развитии и умственная отсталость легкой или средней степени тяжести. Другими проявлениями являются судорожные припадки (55%), врожденные пороки сердца (39%), почечные и урологические аномалии (37%) и крипторхизм (71% мужчин).
Дупликация 17 q 21.31. Реципрокная дупликация обнаруживается у пациентов с серьезным отставанием в психомоторном развитии, микроцефалией, лицевыми дисморфизмами, аномальными пальцами и гирсутизмом.
Генетические маркеры:
MAPT
KANSL1
Синдром Фелана-МакДермида вызван делецией (терминальной или интерстициальной) или несбалансированной транслокацией участка длинного плеча 22-й хромосомы (22q13.3), включающего в себя критический регион (содержит гены SHANK3, ACR, RABL2B).
Характеризуется неонатальной гипотонией, отставанием в развитии умеренной или тяжелой степени, нарушением речевого развития. Другими проявлениями заболевания являются большие кисти рук, дисплазия ногтей на ногах и сниженное потоотделение, что может привести к гипертермии. Еще одна особенность поведения, которую демонстрируют более чем 80 процентов детей, - жевание/облизывание несъедобных предметов. Кроме этого наблюдается сниженный болевой порог и аутичноподобные проявления.
В половине случаев мутация возникает de novo в процессе гаметогенеза (чаще - сперматогенеза). В остальных случаях мутация (несбалансированная транслокация) возникает вследствие передачи генетического материала от родителя, несущего сбалансированную транслокацию. При этом риск развития заболевания у сибсов значительно возрастает, в связи с чем показано генетическое обследование родителей.
.Генетические маркеры:
SHANK3
RABL2B
Синдром дупликации гена MECP 2 - тяжелое неврологическое расстройство, характеризующееся младенческой гипотонией, отставанием в психомоторном и умственном развитии, прогрессирующей спастичностью, рецидивирующими респираторными заболеваниями (примерно у 75% больных) и судорожными припадками (примерно в 50% случаев). Синдром дупликации MECP2 имеет 100% пенетрантность у мужчин. У женщин с дупликацией MECP2 гена симптоматика наблюдается при сопутствующих аномалиях Х-хромосом, которые предотвращают инактивацию дуплицированного участка. Генерализованные тоническо-клонические припадки наблюдаются наиболее часто. Треть пациентов-мужчин неспособны независимо передвигаться. Почти 50% пациентов-мужчин умирают до 25 лет от осложнений рецидивирующих инфекций и/или ухудшения неврологического статуса. Помимо основных проявлений, наблюдаются аутичные черты поведения и гастроинтестинальная дисфункция.
Генетические маркеры:
MECP2
Е.В. Тозлиян, педиатр-эндокринолог, генетик, к. м. н., Обособленное структурное подразделение «Научно-исследовательский клинический институт педиатрии» ГБОУ ВПО РНИМУ им. Н.И. Пирогова Минздрава РФ, г. Москва Ключевые слова
: дети, синдром Нунан, диагностика.
Key words
: children, syndrome Noonan, diagnostics.
В статье описан синдром Нунан (синдром Ульриха – Нунан, тернероидный синдром с нормальным кариотипом) – редкая врожденная патология, наследуется по аутосомно-доминантному типу, носит семейный характер, однако встречаются и спорадические случаи. Синдром предполагает наличие фенотипа, характерного для синдрома Шерешевского – Тернера у особей женского и мужского пола с нормальным кариотипом. Представлено клиническое наблюдение. Показаны сложности дифференциальнодиагностического поиска, недостаточная информированность клиницистов о данном синдроме и важность междисциплинарного подхода.
Исторические факты
Впервые о необычном синдроме упомянул О. Kobylinski в 1883 году (фото 1).
Старейший известный клинический случай синдрома Нунан, описан в 1883 году О. Kobylinski
Болезнь описана в 1963 году американским врачом-кардиологом Жаклин Нунан, сообщившей о девяти пациентах со стенозом клапана легочной артерии, малым ростом, гипертелоризмом, умеренным снижением интеллекта, птозом, крипторхизмом и скелетными нарушениями. Доктор Нунан, практиковавшая как детский кардиолог в университете Айовы, заметила, что у детей с редким типом порока сердца – стенозом клапана легочной артерии – часто наблюдались типичные физические аномалии в виде низкого роста, крыловидной шеи, широко посаженных глаз и низко расположенных ушей. Мальчики и девочки поражались одинаково. Доктор Джон Опиц, бывший студент Нунан, первым ввел в употребление термин «синдромом Нунан» для характеристики состояния детей, у которых отмечались признаки, похожие на описанные Нунан. Позже Нунан написала статью «Гипертелоризм с фенотипом Тернера», и в 1971 году на симпозиуме сердечнососудистых заболеваний название «синдром Нунан» стало официально признанным .
Этиология и патогенез
Синдром Нунан представляет собой аутосомно-доминантное заболевание с варьирующей экспрессивностью (рис. 1). Ген синдрома Нунан локализован на длинном плече хромосомы 12 . Не исключена генетическая гетерогенность синдрома. Описаны спорадические и семейные формы синдрома с аутосомно-доминантной формой наследования. В семейных случаях мутантный ген наследуется, как правило, от матери, так как из-за тяжелых пороков развития мочеполовой системы мужчины с этим заболеванием часто бесплодны. Большинство описанных случаев являются спорадическими, вызванными мутациями de novo.

. Аутосомно-доминантный тип наследования
Описанные сочетания синдрома Нунан с нейрофиброматозом I типа в нескольких семьях заставило предположить возможную связь двух независимых локусов 17q11.2 хромосомы 17. У некоторых больных выявляются микроделеции в локусе 22q11 хромосомы 22; в этих случаях клинические проявления синдрома Нунан сочетаются с гипофункцией тимуса и синдромом Ди Джорджи. Ряд авторов обсуждают участие в патогенезе синдрома предполагаемых генов лимфогенеза в связи с наличием сходных с синдромом Тернера лицевых и соматических аномалий и высокой частоты патологии лимфатической системы .
Наиболее частая причина синдрома Нунан – мутация гена PTPN11, которая обнаруживается приблизительно у 50% больных. Белок, кодируемый геном PTPN11, относится к семейству молекул, регулирующих ответ эукариотических клеток на внешние сигналы. Наибольшее число мутаций при синдроме Нунан локализовано в экзонах 3,7 и 13 гена PTPN11, кодирующих домены белка, отвечающие за переход протеина в активное состояние .
Возможные представления о патогенезе представлены следующими механизмами:
RAS-MAPK-путь – очень важный путь сигнальной трансдукции, через который внеклеточные лиганды – определенные факторы роста, цитокины и гормоны – стимулируют клеточную пролиферацию, дифференцирование, выживаемость и метаболизм (рис. 2). После связывания лиганда рецепторы на поверхности клеток фосфорилируются в местах их эндоплазматического региона. Это связывание задействует адаптерные протеины (например, GRB2), которые формируют конститутивный комплекс с факторами обмена гуаниновых нуклеотидов (например, SOS), конвертирующих неактивный ГДФ-связанный RAS в его активную ГТФ-связанную форму. Активированные RAS-протеины затем активируют RAF-MEKERKкаскад через ряд реакций фосфорилирования. В результате активированный ERK проникает в ядро для изменения транскрипции целевых генов и корректирует активность эндоплазматических мишеней для индукции адекватных кратковременных и длительных клеточных ответов на стимул. Все гены, вовлеченные в синдром Нунан, кодируют интегральные для этого пути протеины, и мутации, вызывающие болезнь, обычно усиливают сигнал, проходящий через этот путь.

. RAS-MAPK-сигнальный путь. Ростовые сигналы передаются с активированных фактором роста рецепторов к ядру. Мутации в PTPN11, KRAS, SOS1, NRAS и RAF1 ассоциированы с синдромом Нунан, а мутации в SHOC2 и CBL ассоциированы с подобным синдрому Нунан фенотипом
Клиническая характеристика синдрома Нунан
Фенотип больных с синдромом Нунан напоминает синдром Тернера: короткая шея с крыловидной складкой или низким ростом волос, низкий рост, гипертелоризм глазных щелей (фото 2). Лицевые микроаномалии включают антимонголоидный разрез глазных щелей, опущенные вниз наружные углы глазных щелей, птоз, эпикантус, низко расположенные ушные раковины, складчатый завиток ушных раковин, аномалии прикуса, расщелину язычка мягкого неба, готическое небо, микрогнатию и микрогению. Грудная клетка щитовидной формы с гипоплазированными широко расставленными сосками, грудина выступает в верхней части и западает в нижней. Около 20% больных имеют умеренно выраженную патологию скелета. Наиболее часто встречаются воронкообразная деформация грудной клетки, кифоз, сколиоз; реже – уменьшение числа шейных позвонков и их сращение, напоминающее аномалии при синдроме Клиппеля – Фейля .

. Фенотипы синдрома Нунан
У больных с синдромом Нунан обычно светлые густые вьющиеся волосы с необычным ростом на темени, часто встречаются пигментные пятна на коже, гипертрихоз, дистрофия ногтевых пластинок, аномалии прорезывания и расположения зубов, склонность к образованию келоидных рубцов, повышенная растяжимость кожи. У трети больных отмечаются периферические лимфатические отеки, чаще лимфедема кистей и стоп проявляется у детей раннего возраста. Нередким признаком является патология зрения (миопия, косоглазие, умеренный экзофтальм и др.). Задержка роста встречается примерно у 75% больных, больше выражена у мальчиков и обычно незначительна. Отставание в росте манифестирует в первые годы жизни, реже отмечается незначительный дефицит роста и массы при рождении. С первых месяцев жизни отмечается снижение аппетита. Костный возраст обычно отстает от паспортного.
Характерным признаком синдрома является одно- или двусторонний крипторхизм, встречающийся у 70–75% больных мужского пола, у взрослых больных отмечается азооспермия, олигоспермия, дегенеративные изменения яичек. Тем не менее пубертат наступает спонтанно, иногда с некоторой задержкой. У девочек часто отмечается задержка становления менструации, иногда – нарушения менструального цикла. Фертильность может быть нормальной у больных обоих полов.
Умственная отсталость выявляется более чем у половины больных, как правило, незначительная. Часто отмечаются особенности поведения, расторможенность, синдром дефицита внимания. Речь обычно развита лучше, чем другие интеллектуальные сферы. Степень снижения интеллекта не коррелирует с тяжестью соматических нарушений [Маринчева Г.С., 1988]. В единичных случаях описываются пороки развития центральной нервной системы (гидроцефалия, спинномозговые грыжи), тромбоэмболические инфаркты мозга, возможно, связанные с гипоплазией сосудов .
Пороки внутренних органов при синдроме Нунан достаточно характерны. Наиболее типичными являются сердечно-сосудистые аномалии: клапанный стеноз легочной артерии (около 60% больных), гипертрофическая кардиомиопатия (20–30%), структурные аномалии митрального клапана, дефекты предсердной перегородки, тетрада Фалло; коарктация аорты описана только у больных мужского пола.
У трети больных регистрируются пороки мочевыделительной системы (гипоплазия почек, удвоение лоханок, гидронефроз, мегауретер и др.).
Достаточно часто при синдроме Нунан отмечается повышенная кровоточивость, особенно при оперативных вмешательствах в ротовой полости и носоглотке. Обнаруживаются различные дефекты коагуляции: недостаточность тромбоцитарной системы, снижение уровня факторов свертывания, особенно XI и XII, увеличение тромбопластинового времени . Имеются сообщения о сочетании синдрома Нунан с лейкемией и рабдомиосаркомой, что может свидетельствовать о некотором повышении риска малигнизации у этих больных .
В таблице 1 представлены особенности фенотипа при синдроме Нунан, меняющиеся с возрастом пациента. В таблице 2 – корреляция между фенотипом и генотипом при синдроме Нунан.
Таблица 1 . Типичные черты лица у больных синдромом Нунан по возрастам
| Лоб, лицо, волосы | Глаза | Уши | Нос | Рот | Шея | |
| Новорожденный* | Высокий лоб, низкая линия роста волос в затылочной области | Гипертелоризм, наклонные книзу глазные щели, складка эпикантуса | – | Короткий и широкий утопленный корень, вздернутый кончик | Глубоко утопленный губной желобок, высокие широкие пики красной каймы губ, микрогнатия | Избыточная кожа на затылке |
| Грудной (2–12 мес.) | Большая голова, высокий и выпирающий лоб | Гипертелоризм, птоз или толстые нависающие веки | – | Короткий и широкий утопленный корень | – | – |
| Ребенок (1–12 лет) | Грубые черты, вытянутое лицо | – | – | – | – | – |
| Подросток (12–18 лет) | Миопатичес-кое лицо | – | – | Мостик высокий и тонкий | – | Очевидное формирование шейных складок |
| Взрослый (>18 лет) | Отличительные черты лица утонченные, кожа кажется тонкой и прозрачной | – | – | Выпирающая носогубная складка | – | – |
| Все возрасты | – | Голубые и зеленые радужные оболочки, ромбовидные брови | Низкие, ротированные назад уши с толстыми складками | – | – | – |
Таблица 2 . Корреляции между генотипом и фенотипом при синдроме Нунан*
| Сердечнососудистая система | Рост | Развитие | Кожа и волосы | Другое | |
| PTPN11 (примерно 50%) | Более выражен стеноз легочного ствола; меньше – гипертрофическая кардиомиопатия и дефект межпредсердной перегородки | Более низкий рост; ниже концентрация IGF1 | Пациенты с N308D и N308S имеют слабое снижение или нормальный интеллект | – | Больше выражен геморрагический диатез и ювенильная миеломоноцитарная лейкемия |
| SOS1 (примерно 10%) | Меньше дефект межпредсердной перегородки | Более высокий рост | Меньше снижение интеллекта, задержка развития речи | Подобны сердечно-кожно-лицевому синдрому | – |
| RAF1 (примерно 10%) | Больше тяжелая гипертрофическая кардио-миопатия | – | – | Больше родимых пятен, лентиго, пятен кофе с молоком | – |
| KRAS (<2%) | – | – | Более тяжелая задержка когнитивного развития | Подобны сер-дечно-кожно-лицевому синдрому | – |
| NRAS (<1%) | – | – | – | – | – |
Данные лабораторных и функциональных исследований
Специфических биохимических маркеров для диагностики синдрома Нунан не существует. У некоторых больных выявляется снижение спонтанной ночной секреции гормона роста при нормальном ответе на фармакологические стимулирующие тесты (клофелином и аргинином), снижение уровня соматомедина-С и снижение реакции соматомединов на введение гормона роста.
Критерии диагноза
Диагноз «синдром Нунан» ставится на основании клинических признаков, в некоторых случаях диагноз подтверждается результатами молекулярно-генетического исследования. Критерии диагностики синдрома включают наличие характерного лица (при нормальном кариотипе) в сочетании с одним из следующих признаков: патологии сердца, низкий рост или крипторхизм (у мальчиков), задержка полового созревания (у девочек). Для выявления сердечно-сосудистой патологии необходимо проведение ультразвукового исследования сердца с динамическим определением размеров полостей и стенки желудочков. Возможна пренатальная диагностика заболевания при помощи ультразвукового мониторинга, позволяющего выявить пороки сердца и аномалии строения шеи .
Дифференциальная диагностика
У девочек дифференциальный диагноз проводится в первую очередь с синдромом Тернера; уточнить диагноз позволяет цитогенетическое исследование. Фенотипические признаки синдрома Нунан встречаются при ряде других заболеваний: синдроме Вильямса, синдроме LEOPARD, Дубовица, кардиофацио-кожном синдроме, Корнелии де Ланге, Коэна, Рубинштейна – Тейби и др. Точная идентификация этих заболеваний будет возможна только при проведении молекулярногенетических исследований каждого синдрома при значительном клиническом материале, что в настоящее время активно развивается.
Лечение
Лечение больных с синдромом Нунан направлено на устранение пороков сердечно-сосудистой системы, нормализацию психических функций, стимуляцию роста и полового развития. Для лечения больных с дисплазией клапанов легочной артерии, помимо прочих методов, с успехом применяется баллонная вальвулопластика. С целью стимуляции психического развития применяются ноотропные и сосудистые средства. Препараты, направленные на стимуляцию полового развития, показаны в основном больным с крипторхизмом. Применяются препараты хорионического гонадотропина в возрастных дозировках. В старшем возрасте – при наличии гипогонадизма – препараты тестостерона. В последние годы применяются рекомбинантные формы гормона роста человека в лечении больных с синдромом Нунан . Клинические данные подтверждаются увеличением на фоне терапии уровня соматомедина-С и специфического связывающего белка. Конечный рост больных, длительное время получающих терапию гормоном роста, в некоторых случаях превышает средний рост членов семьи.
Прогноз для жизни определяется тяжестью сердечно-сосудистой патологии.
Профилактика болезни основывается на данных медико-генетического консультирования.
Медико-генетическое консультирование
При медико-генетическом консультировании следует исходить из аутосомно-доминантного типа наследования и высокого (50%) риска повторения заболевания в семье при унаследованных формах. С целью идентификации характера типа наследования необходимо проводить тщательное обследование родителей, так как синдром может проявляться минимальными клиническими симптомами. В настоящее время разработана и совершенствуется молекулярно-генетическая диагностика заболевания путем типирования мутаций в генах: PTPN11, SOS1, RAF1, KRAS, NRAS и др. Разрабатываются способы пренатальной диагностики заболевания.
Клиническое наблюдение
Мальчик Г., 9 лет (фото 3), наблюдался по месту жительства врачом-генетиком с диагнозом «хромосомная патология?, синдром Вильямса (своеобразный фенотип, уплотнение створок митрального клапана, гиперкальциемия однократно в 3 года)?.

. Особенности фенотипа ребенка с синдромом Нунан (удлиненный лицевой скелет с «пухлыми щечками», короткая шея, крыловидные складки на шее, нос укорочен с открытыми вперед ноздрями, пухлые губы, скошенный подбородок, антимонголоидный разрез глазных щелей, неправильный прикус, макростомия)
Жалобы на сниженную память, утомляемость, сниженные темпы роста.
Анамнез семейный : родители русские по национальности, не состоящие в кровном родстве и не имеющие профессиональных вредностей, здоровые. Рост отца – 192 см, рост матери – 172 см. В родословной случаев психических заболеваний, эпилепсии, задержки в развитии не отмечалось.
Анамнез жизни и заболевания : мальчик от 2-й беременности (1-я беременность – м/а), протекавшей с угрозой прерывания на всем протяжении, сопровождающейся многоводием. Роды первые, в срок, стремительные, масса при рождении – 3400 г, длина – 50 см. Закричал сразу, оценка по шкале Апгар – 7/9 баллов. При рождении неонатологом обращено внимание на необычный фенотип ребенка, рекомендовано исследование кариотипа, результат – 46, XY (нормальный мужской кариотип). Был заподозрен врожденный гипотиреоз, проведено исследование тиреоидного профиля, результат – нормальный тиреоидный статус. Далее ребенок наблюдался генетиком с предполагаемым диагнозом «синдром Вильямса». Ранний постнатальный период – без особенностей. Моторное развитие по возрасту, первые слова – к году, фразовая речь – в 2 года 3 мес.
В возрасте 8 лет консультирован эндокринологом по поводу сниженных темпов роста, утомляемости, сниженной памяти. При рентгенологическом исследовании кистей рук выявлено умеренное отставание костного возраста (КВ) от паспортного (КВ соответствовал 6 годам). При исследовании тиреоидного профиля выявлено умеренное повышение тиреотропного гормона при нормальном уровне свободного Т4 и остальных показателей; УЗИ щитовидной железы – без патологии. Назначена гормональная терапия с последующим динамическим наблюдением.
Учитывая неопределенность диагноза по месту жительства, генетиком ребенок направлен в Московский областной консультативно-диагностический центр для детей с целью уточнения диагноза.
Данные объективного исследования:
Рост – 126 см, вес – 21 кг.
Физическое развитие ниже среднего, гармоничное. Sds роста соответствует –1 (норма – –2+2). Особенности фенотипа (фото 3): удлиненный лицевой скелет с «пухлыми щечками», короткая шея, крыловидные складки на шее, низкий рост волос на шее, нос укороченный с открытыми вперед ноздрями, пухлые губы, скошенный подбородок, антимонголоидный разрез глазных щелей, неправильный прикус, макростомия, гипертелоризм сосков, асимметрия грудной клетки, на стопах неполная кожная синдактилия 2–3-го пальцев, выраженная гипермобильность межфаланговых суставов, ломкие, сухие ногти. По внутренним органам – без особенностей. Половое развитие – Tanner I (что соответствует допубертатному периоду).
Данные лабораторных и функциональных исследований:
Клинический анализ крови и мочи – норма.
Биохимический анализ крови – показатели в пределах нормы.
Тиреоидный профиль (ТТГ) – 7,5 мкМЕ/ мл (норма – 0,4–4,0), остальные показатели в норме.
Соматотропный гормон (СТГ) – 7 нг/мл (норма – 7–10), соматомедин-С – 250 нг/мл (норма – 88–360).
УЗИ щитовидной железы – без патологии.
УЗИ внутренних органов – без особенностей.
ЭКГ – синусовая тахикардия, нормальное положение электрической оси сердца.
ЭхоКГ – ПМК I степени с минимальной регургитацией, миксоматозное утолщение створок митрального клапана, дополнительная хорда в полости левого желудочка.
R-графия позвоночника – правосторонний сколиоз грудного отдела позвоночника I степени.
R-графия кистей рук с захватом предплечий – костный возраст 7–8 лет.
ЭЭГ-паттернов эпилептической активности не зарегистрировано.
МРТ головного мозга – без патологических изменений.
Аудиограмма – без патологии.
ДНК-диагностика: молекулярно-генетическое исследование – делеций исследуемых локусов критического района хромосомы 7 не выявлено; обнаружена мутация Gly434Ary (1230G>A) в 11-м экзоне гена SOS1 (анализ гена PTPN11 – мутаций не обнаружено), что характерно для синдрома Нунан.
Консультации специалистов:
Эндокринолог – субклинический гипотиреоз, неполная медикаментозная компенсация.
Окулист – астигматизм.
Невролог – вегетососудистая дистония. Невротические реакции.
Кардиолог – функциональная кардиопатия.
Хирург-ортопед – нарушение осанки. Деформация грудной клетки.
Генетик – синдром Нунан.
Учитывая фенотип ребенка, данные анамнеза, результаты дополнительных исследований, поставлен диагноз «синдром Нунан», что подтверждено результатом молекулярно-генетического исследования.
Таким образом, представленное клиническое наблюдение демонстрирует сложности дифференциально-диагностического поиска, необходимость интегрировать отдельные признаки в общий фенотип того или иного патологического состояния для целенаправленной своевременной диагностики отдельных форм наследственных заболеваний, важность молекулярно-генетических методов для уточнения диагноза. Своевременная диагностика, уточнение генеза каждого синдрома особенно важны, так как позволяют найти оптимальный подход к лечению этих состояний, профилактике возможных осложнений (вплоть до инвалидности ребенка); предупреждению повторного возникновения наследственных болезней в пораженных семьях (медико-генетическое консультирование). Это диктует необходимость врачам различных специальностей четко ориентироваться в потоке наследственно обусловленной патологии.
Список литературы:
- Baird P., De Jong B. Noonan’s syndrome (XX and XY Turner phenotype) in three generations of a family // J. Pediatr., 1972, vol. 80, p. 110–114.
- Hasegawa T., Ogata T. et al. Coarctation of the aorta and renal hupoplasia in a boy with Turner/Noonan surface anomalies and a 46, XY karyotype: a clinical model for the possible impairment of a putative lymphogenic gene(s) for Turner somatic stigmata // Hum. Genet., 1996, vol. 97, р. 564–567.
- Федотова Т.В., Кадникова В.А. и соавт. Клинико-молекулярно-генетический анализ синдрома Нунан. Материалы VI съезда Российского общества медицинских генетиков. Медицинская генетика, приложение к № 5, 2010, с.184.
- Ward K.A., Moss C., McKeown C. The cardio-facio-cutaneous syndrome: a manifestation of the Noonan syndrome? // Br. J. Dermatol., 1994, vol. 131, р. 270–274.
- Municchi G., Pasquino A.M. et al. Growth hormone treatment in Noonan syndrome: report of four cases who reached fi nal height // Horm. Res., 1995, vol. 44, р. 164–167.
Детекция мутации denovo в гене дистрофина и её значение для медико-генетического консультирования при мышечной дистрофии Дюшенна
(клиническое наблюдение)
Муравлева Э.А., Стародубова А.В, Пышкина Н.П., Дуйсенова О.С.
Научный руководитель: д.м.н. доц. Колоколов О.В.
ГБОУ ВПО Саратовский ГМУ им. В.И. Разумовского Минздрава РФ
Кафедра неврологии ФПК и ППС им. К.Н. Третьякова
Введение. Мышечная дистрофия Дюшенна (МДД) относится к наиболее часто встречающимся наследственным нервно-мышечным болезням. Распространенность её составляет 2-5: 100000 населения, популяционная частота - 1: 3500 новорожденных мальчиков. Эта форма мышечной дистрофии впервые описана Edward Meryon (1852г.) и Guillaume Duchenne (1861г.).
Заболевание характеризуется Х-сцепленным рецессивным типом наследования и тяжелым, прогрессирующим течением. МДД обусловлена мутацией в гене дистрофина, локус которого локализован на Xp21.2. Около 30% случаев обусловлены мутациями de novo, 70% - носительством мутации матерью пробанда. Дистрофин отвечает за соединение цитоскелета каждого мышечного волокна с основной базальной пластинкой (внеклеточного матрикса) через белковый комплекс, который состоит из многих субъединиц. Отсутствие дистрофина приводит к проникновению избыточного кальция в сарколему (клеточную мембрану). Мышечные волокна подвергаются некрозу, происходит замещение мышечной ткани жировой, а также соединительной.
Современная диагностика МДД основана на оценке соответствия проявлений болезни клинико-анамнестическим и лабораторно-инструментальным (креатин-киназа сыворотки крови (ККС), электронейромиография (ЭНМГ), гистохимическое исследование мышечного биоптата) критериям, генеалогическом анализе и данных молекулярно-генетического исследования.
Проведение медико-генетического консультирования в настоящее время во многих семьях позволяет предупредить рождение больного ребенка. Пренатальная ДНК диагностика на ранних сроках беременности в семьях, имеющих ребенка, страдающего МДД, позволит выбрать дальнейшую тактику для родителей и, возможно, досрочно прекратить беременность в случае наличия заболевания у плода.
В ряде случаев клиническая картина наблюдается у женщин - гетерозиготных носительниц мутантного гена в виде увеличения икроножных мышц, умеренно выраженной мышечной слабости, снижения сухожильных и периостальных рефлексов, по данным параклинических исследований повышается уровень ККС. Кроме того, классические клинические проявления МДД могут возникать у женщин с синдромом Шерешевского-Тернера (генотип 45, ХО).
Клинический пример. В нашей клинике наблюдается мальчик К., 7 лет, который предъявляет жалобы на слабость в мышцах рук и ног, утомляемость при длительной ходьбе. Мама ребенка отмечает у него периодические падения, затруднения при подъеме по лестнице, нарушение походки (по типу «утиной»), трудности при вставании из положения сидя, увеличение икроножных мышц в объеме.
Раннее развитие ребенка протекало без особенностей. В возрасте 3-х лет окружающие заметили нарушения двигательных функций в виде появления трудностей при ходьбе по лестнице, при вставании, ребенок не принимал участия в подвижных играх, стал быстро уставать. Затем изменилась походка по типу «утиной». Наросли трудности при вставании из положения сидя или из положения лежа: поэтапное вставание «лесенкой» с активным использованием рук. Постепенно стало заметным увеличение икроножных и некоторых других мышц в объеме.
В неврологическом осмотре ведущим клиническим признаком является симметричный проксимальный периферический тетрапарез, более выраженный в ногах (мышечная сила в проксимальных отделах верхних конечностей - 3-4 балла, в дистальных - 4 балла, в проксимальных отделах нижних конечностей - 2-3 балла, в дистальных - 4 балла). Походка изменена по типу «утиной». Использует вспомогательные («миопатические») приемы, например вставание «лесенкой». Мышечный тонус снижен, контрактур нет. Гипотрофия мышц тазового и плечевого пояса. «Миопатические» черты, например в виде широкого межлопаточного пространства. Имеется псевдогипертрофия икроножных мышц. Сухожильные и периостальные рефлексы - без достоверной разницы сторон; биципитальные - низкие, триципитальные и карпорадиальные - средней живости, коленные и ахилловы - низкие. На основании клинических данных заподозрена МДД.
При исследовании ККС её уровень составил 5379 ед/л, что в 31 раз выше нормы (норма - до 171 ед/л). По данным ЭНМГ зарегистрированы признаки, более характерные для умеренно текущего первично-мышечного процесса. Таким образом, полученные данные подтвердили наличие у пациента МДД.
Помимо пробанда осмотрены его родители и старшая родная сестра. Ни у кого из родственников пробанда клинических проявлений МДД не наблюдалось. Однако у матери замечено некоторое увеличение икроножных мышц в объеме. По данным генеалогического анализа пробанд является единственным заболевшим в семье. При этом нельзя исключить, что мать ребенка и родная сестра пробанда являются гетерозиготными носительницами мутантного гена (рис. 1).
Рис. 1 Родословная
В рамках медико-генетического консультирования семья К. была обследована на предмет наличия/отсутствия делеций и дупликаций в гене дистрофина. Молекулярно-генетический анализ в лаборатории ДНК-диагностики МГНЦ РАМН выявил у пробанда К. делецию 45 экзона, что окончательно подтверждает установленный клинический диагноз МДД. У матери делеция 45 экзона, выявленная у сына, не обнаружена. У сестры в результате анализа делеция 45 экзона, выявленная у брата, не найдена. Следовательно, у исследуемого мутация, скорее всего, имеет происхождение de nоvo, однако также она может явиться результатом герминального мозаицизма у матери. Соответственно, при мутации de novo риск рождения больного ребенка у матери будет определяться популяционной частотой данной мутации (1:3500, ‹‹1%), что значительно меньше, нежели при Х-сцепленном рецессивном типе наследования (50% мальчиков). Поскольку невозможно полностью исключить, что мутация может явиться результатом герминального мозаицизма, при котором наследование по законам Менделя нарушается, рекомендуется проведение пренатальной диагностики при последующей беременности у матери и сестры пробанда.
Заключение. В настоящее время у врача есть широкий арсенал симптоматических средств, используемых в лечении МДД, однако, несмотря на достижения науки, этиологическое лечение МДД до сих пор не разработано, эффективных препаратов для заместительного лечения при МДД не существует. Согласно недавним исследованиям стволовых клеток, существуют перспективные векторы, которые могут заменить поврежденные мышечные ткани. Однако, в настоящее время, возможно лишь симптоматическое лечение, направленное на улучшение качества жизни больного. В этой связи ранняя диагностика МДД играет важнейшую роль для своевременного проведения медико-генетического консультирования и выбора дальнейшей тактики планирования семьи. Для пренатальной ДНК диагностики исследование с помощью биопсии хориона (CVS) можно проводить на 11-14 неделях беременности, амниоцентез можно использовать после 15 недели, забор крови плода возможен примерно на 18 неделе. Если тестирование будет осуществлено на ранних сроках беременности, возможно досрочное прекращение беременности в случае наличия заболевания у плода. В ряде случаев целесообразно проведение преимплантационной ДНК диагностики с последующим экстракорпоральным оплодотворением.
Выводы. Для обеспечения раннего выявления и профилактики МДД необходимо шире использовать методы молекулярно-генетической диагностики; повысить настороженность практикующих врачей в отношении данной патологии. При мутации de novo риск рождения больного ребенка у матери определяется популяционной частотой мутации гена дистрофина. В случаях носительства мутации матерью пробанда требуется пренатальная или перимплантационная ДНК диагностика с целью планирования семьи.
Похожие статьи
-
Мытарства души после смерти: что происходит после смерти
Понимание посмертной жизни души очень важно для каждого верующего религиозного человека. Ответив на вопрос, что нас ждет после смерти, что такое душа, мы понимаем, что такое человек и как нужно жить, чтобы не погибнуть для вечности....
-
Штомпка анализ современных обществ
Теория структурации Э. Гидденса послужила в определенной мере толчком для появления в 1990-х гг. работ польского социолога Петра Штомпки (ныне президента Международной социологической ассоциации), посвященных комплексному и целостному...
-
Поиск презентаций. это будет их проект
Презентация: Творческий проект с использованием ученика 1-5 класса МОУ Гимназии 26 Девяткина Дмитрия «Правила поведения младшего школьника при чрезвычайных ситуациях.» Творческий проект с использованием ученика 1-5 класса МОУ Гимназии 26...
-
Когда наступает Новый год Свиньи по китайскому календарю?
Восточная культура и китайские традиции прочно прижились в нашей повседневной жизни, стали и нашими привычками и традициями. Праздновать Новый год по-восточному сегодня стали многие люди, другие же хоть и не празднуют, но какое животное...
-
Сочинение по картине К.Ф.Юона На тему: «Весенний солнечный день. Описание картины К. Юона "Весенний солнечный день" Весенний солнечный день небо
К. Ф. Юон является замечательным и талантливым мастером живописи, которому удалось создать множество примечательных картин. Особое внимание уделялось художником написанию природных особенностей родного края, которые изображены на его...
-
Крымский гуманитарный университет (КГУ)
г.Ялта, пгт. Массандра, ул. Стахановская, 11 Становление и развитие современной кафедры педагогики и управления учебными заведениями начинается с деятельности цикловой комиссии при Ялтинском педагогическом училище. В 1994 году одновременно...